
Sequin Submission to Genbank Tutorial
James A. Fellows Yates
As I came to the end of my M.Sc. and writing the corresponding journal article, one thing I was aware of was that all previous papers on woolly mammoth mitchondrial genomes to NCBI’s GenBank database. To continue with this established ‘tradition’, I decided to investigate how to do this. Worringly, most people I spoke to didn’t have very positive views on this procdure, saying that it was complicated, error prone and badly documented and that it wasn’t worth my time. In fact, with a bit of help from the authors of some of the previous papers and with NCBI itself, the overall task ended up being easier than expected (admittedly after a considerable bit of trial and error).
Here, I describe the eventual steps that I did to (successfully!) annotate a set of mtGenomes and make a submission to GenBank. However, note that these instructions are only valid for this use-case, and would not be necessarily valid for other situations.
In this case, we assume that you have multiple sequences of a species that already has an annotated reference sequence on GenBank. I use here the ‘Krause’ reference (Accession: NC_007596.2) and my reconstructed mtGenomes.
Preparation
- Download the program
Sequinfrom here (If running Ubuntu 16.04 might have to install libxp6 from the PPA here). - Download the reference sequence from Genbank in FASTA format and the FeatureTable format.
- Using a multisequence aligner tool like MEGA or Geneious, align your sequences to the reference, and export the file as a FASTA file (make sure to export gaps as Ns, and not ‘-‘ as in the documentation, and keep the reference sequence!).
-
To save time later, you now want to change the FASTA headers for each sequnce to the NCBI format, adding as much information as you can using the ‘modifiers’ listed here. An example of one of my headers is as so:
>JK2760 [organism=Mammuthus primigenius] [country=Germany] [isolate=JK2760] Mammuthus primigenius isolate JK2760 mitochondrion, partial genome
For the reference sequence you should also add the tag [acc=NC_007596.2].
Using Sequin
- Next we can start
Sequin, select ‘NCBI GenBank’ and ‘Start new submission’.
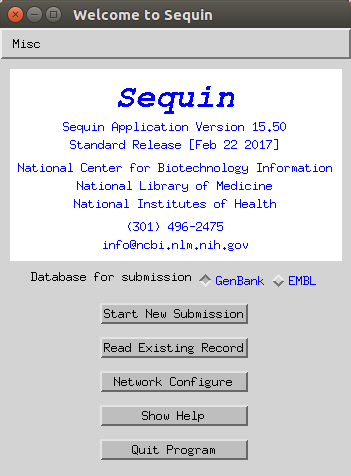
- Set the release date of the data (for example if you want to release on the same day as your paper is published - note you can update this later) and add a tentative title for the study.
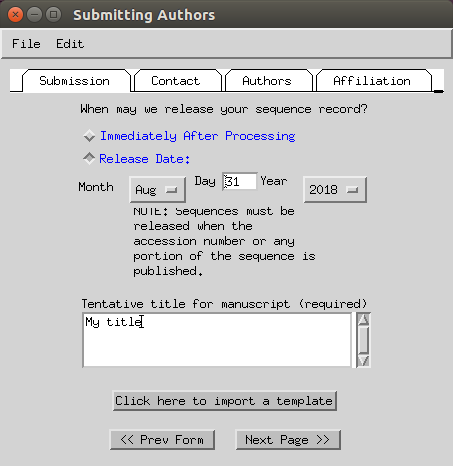
- Set you name, phone and email (fax can be skipped).
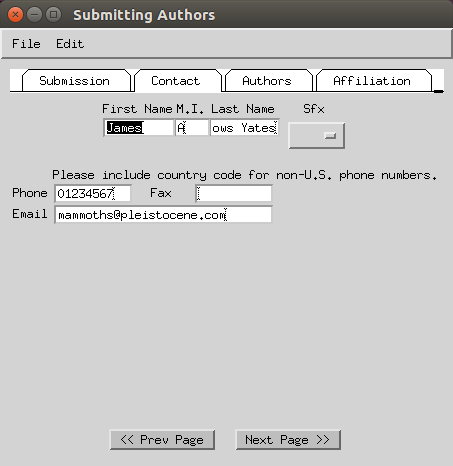
- Enter the names of the authors of the study.
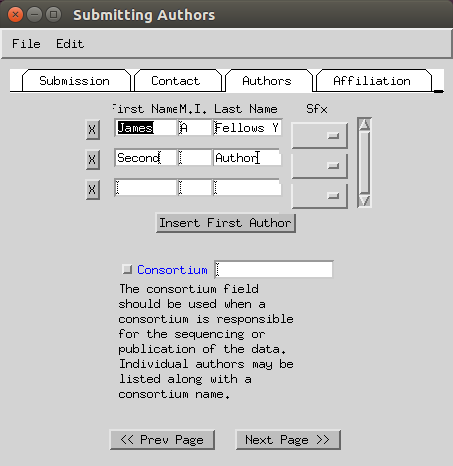
- Enter the submitting institution address (fax can be skipped).
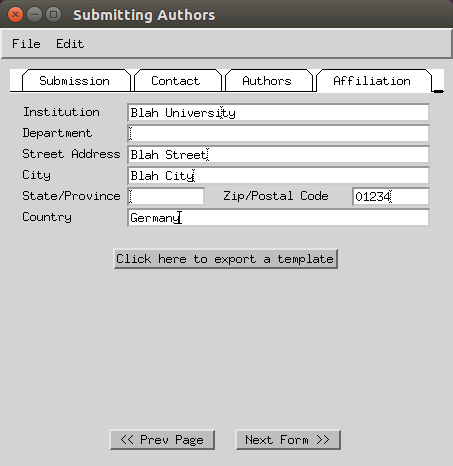
-
Select the ‘use normal submission dialog’ option.
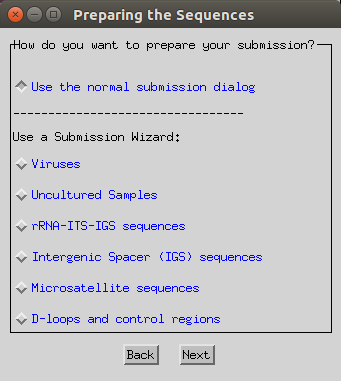
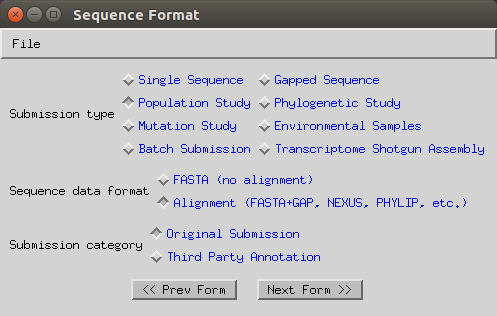
-
Select ‘Population Study’, ‘Alignment’ and ‘Original submission’.
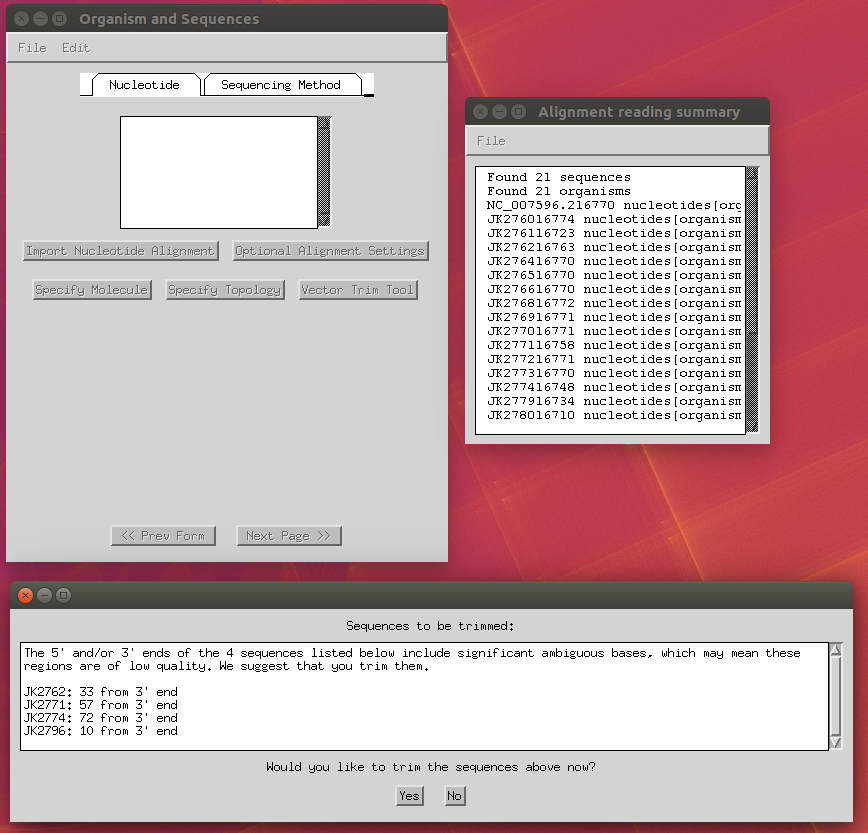
-
Now you can import your FASTA alignment through the ‘Import Alignment’ button and select the following options:
- Trim Ns at the end of sequences: yes.
- Specify molecule: ‘Genomic DNA’ (apply to all).
- Specify topology: ‘linear’ (apply to all).
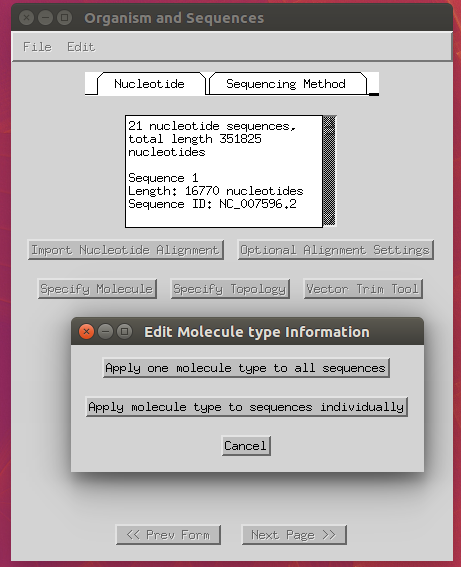
-
Select ‘Illumina’, ‘assembled sequences’ and input your assembly program (in my case it is
bwaand version ‘0.7’).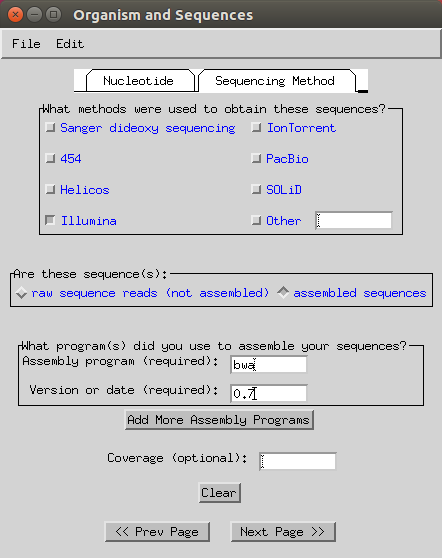
-
Next you can add various information like organisms, locations (e.g. here I selected mitochondrion) and genetic codes (to apply to all you can select the header). You can keep the genetic code as ‘standard’ or change to ‘Vertebrate Mitochondrial’. Some of this will have already been imported based on your FASTA headers.
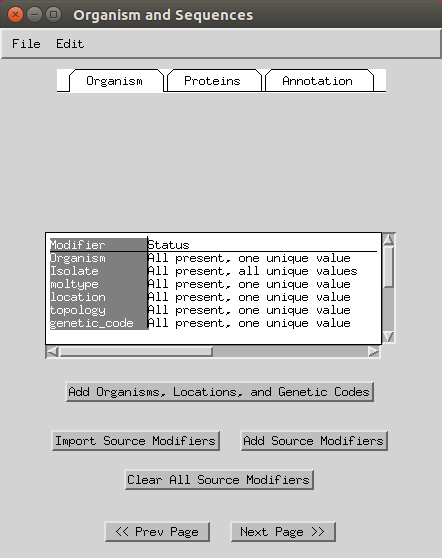
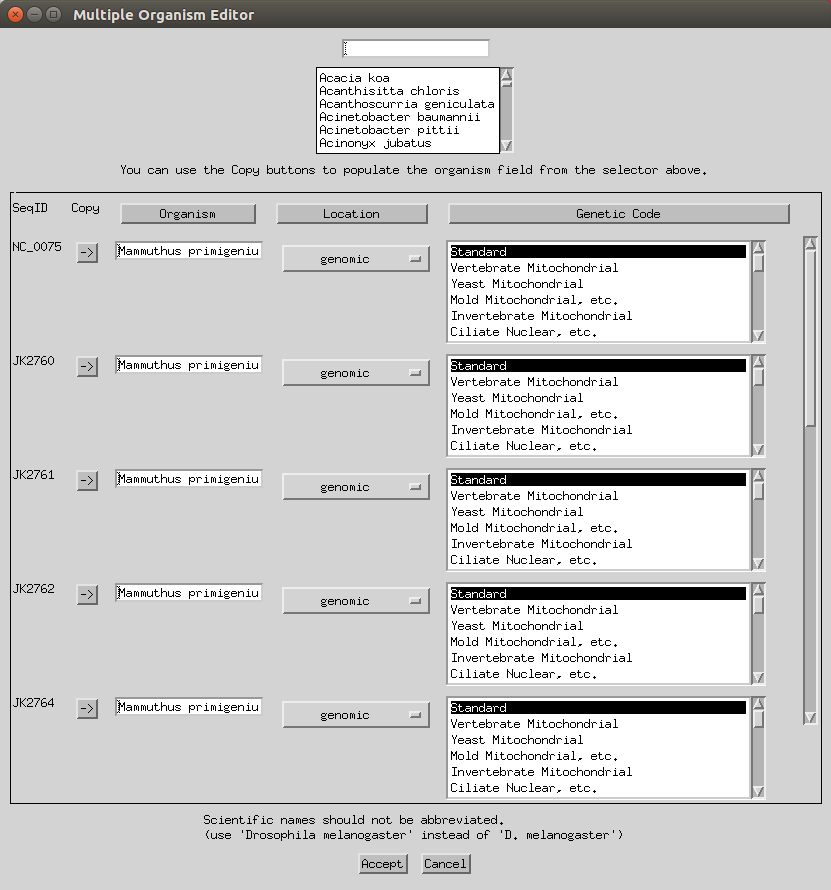
- You can skip the protein step.
-
You can also skip annotation (select ‘none’) and ‘Continue to record viewer’.
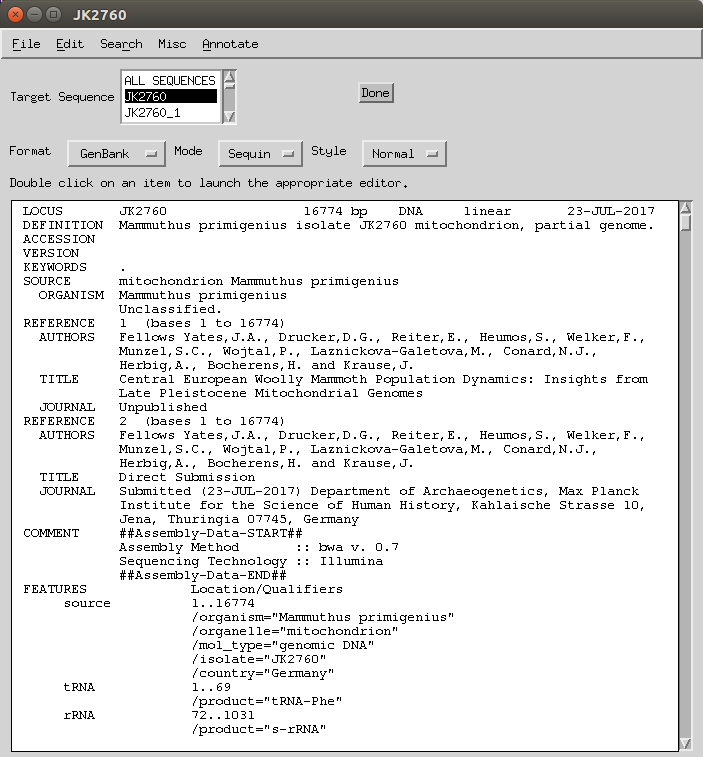
-
First, it is best to annotate any runs of Ns as sequencing gaps (as per advice from the NCBI helpdesk). You can do this by doing the following:
- In record viewer Go to Search Validate > Validate.
-
Double click on a ‘run of Ns’ error.
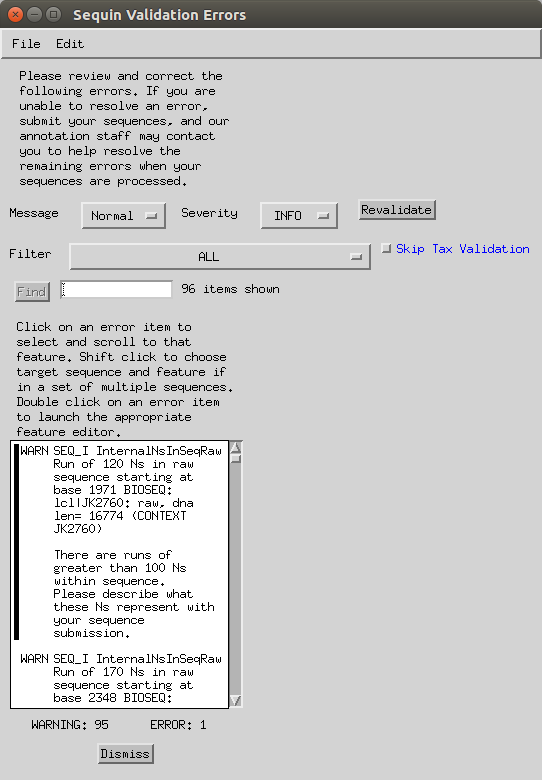
- In the sequence viewer, type in the start position (-1) in the ‘Go to’ box.
-
Then click and drag until all the Ns are selected.
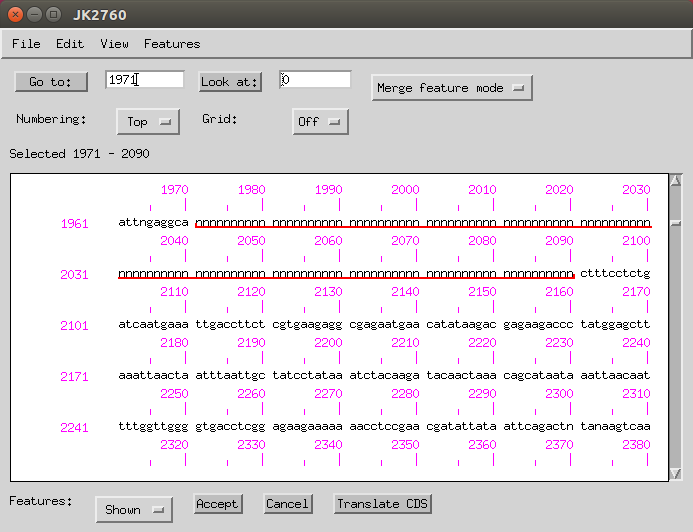
-
Then go to Features > Remaining Features > misc_features and under the ‘Properties’ and then ‘Comment’ tab you can add ‘sequencing gap, estimated length 119 bp’.
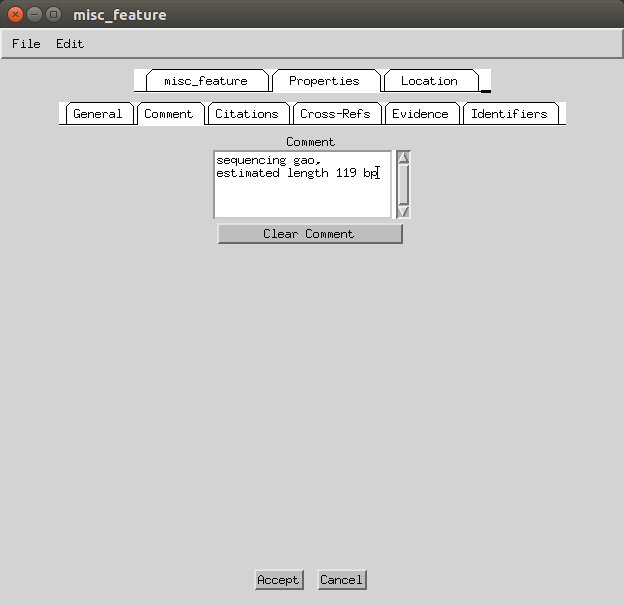
- Once all of those are done, in the Record Viewer you need to select from the drop down menu the reference sequence (in this case NC_007596.2) and then File > Open the Genbank (flat)file or the feature table. This should automatically load all the annotations of the original sequence, also ‘importing’ features that in the latest version of Sequin fail due to conflicts (e.g. I could not manually make a
/trans_exep, but when I imported it gives an error but keeps the feature). -
Finally in the Record Viewer you can go Edit > Feature Propagate. This will copy over all of the annotations on the reference sequnce to your other sequences, but doing the correct DNA to amino acid translation.
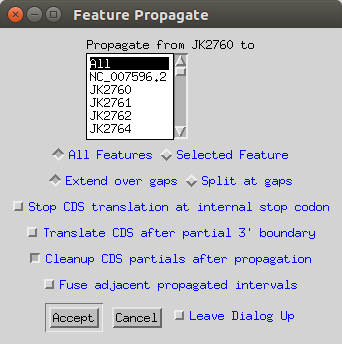
- You should now do another ‘validate’ to find any errors. For example a couple of my translated amino acid sequences began with XXXXXX, so the
Sequincouldn’t find the start codon and thus failed. I just removed these feature entries (and informed NCBI staff). - Once you have no failures, you can go in Record Viewer to ‘File > Prepare Submission’ and send the corresponding file to NCBI genbank as per the instructions. here.
Once done, after a few days you should recieve a message from the NCBI either with corrections to be made or a list of accession numbers, which will go live on the date you selected in Sequin.
To update or to get the release date changed you can just email them again at the same address (you don’t need to do a resubmission or use sequin).